

Пострегистрационный мониторинг медицинских изделий с технологиями искусственного интеллекта является обязательной и важной мерой обеспечения доверия к ИИ-решениям, также служит целям контроля их безопасности. Однако регуляторика в этой сфере довольно сложная и, кроме этого, она в последнее время пересматривается и уточняется. Поэтому мы решили систематизировать текущую систему пострегистрационного мониторинга медизделий с ИИ и рассказать об этом читателям нашего блога.
После регистрации и вывода медицинского изделия (МИ) на рынок должен осуществляться мониторинг его безопасности в соответствии с:
- Решением коллегии Евразийской экономической комиссии от 22.12.2015 №174
- Приказом Министерства здравоохранения РФ от 19 октября 2020 г. N 1113н
Целью мониторинга является выявление и предотвращение побочных действий, неблагоприятных событий, связанных с применением МИ и создающих угрозу жизни и здоровью граждан и медицинских работников.
Процесс включает в себя сбор, обработку, регистрацию и анализ информации о неблагоприятных событиях, в том числе:
- данных о побочных действиях, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия;
- данных о нежелательных реакциях, выявленных при применении медицинских изделий;
- анализ особенностей взаимодействия медицинских изделий между собой;
- фактов и обстоятельств, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации зарегистрированных медицинских изделий.
Классификация неблагоприятных событий, связанных с обращением МИ, утверждена приказом Росздравнадзора №4513 от 20.05.2021. Источником информации могут быть различные каналы, включая получение сведений из государственных информационных систем в сфере здравоохранения, информации от пользователей, данных, предоставленных самим заявителем и т.д
Для всех медицинских изделий класса потенциального риска применения 3, а также медицинских изделий, имплантируемых в организм человека класса потенциального риска применения 26, производитель обязан в течение 3 лет проводить процедуру клинического мониторинга и предоставлять соответствующую отчетность в Федеральную службу по надзору в сфере здравоохранения ежегодно, не позднее 1 февраля.
Схема взаимодействия производителей медицинских изделий с Росздравнадзором в РФ в части пострегистрационного мониторинга представлена ниже.
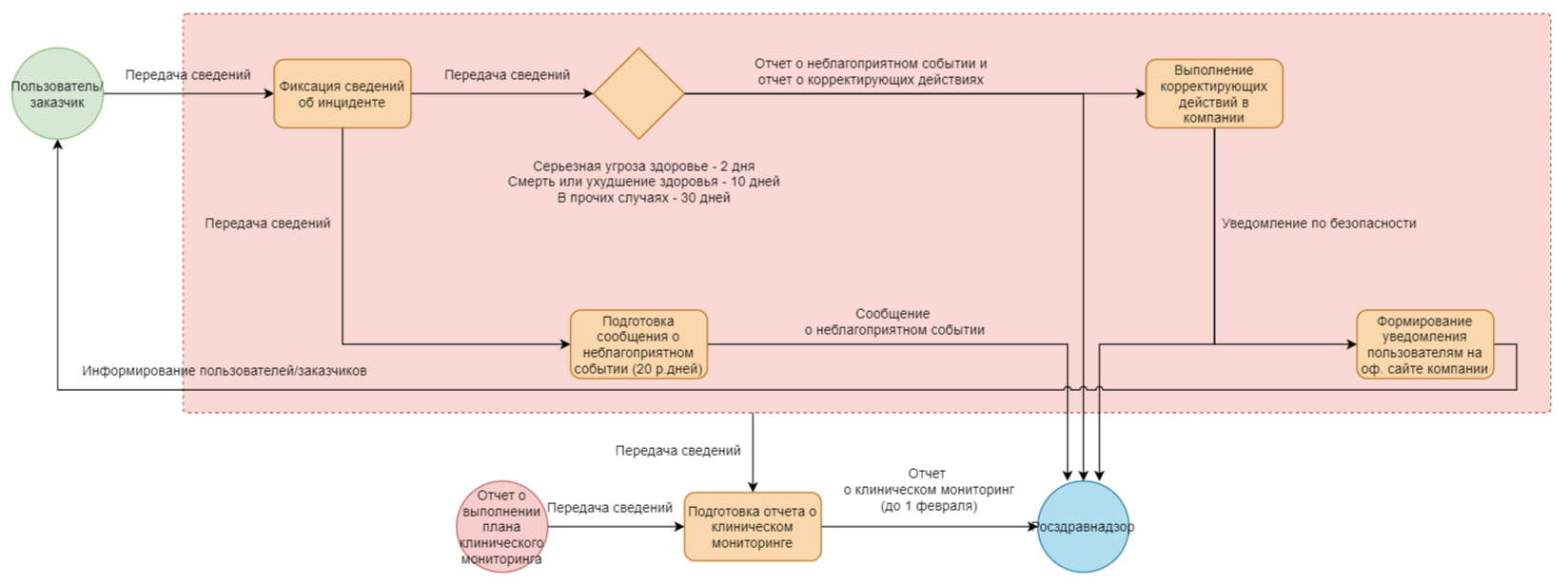
Класс, в зависимости от степени потенциального риска применения в медицинских целях, определяется в соответствии с приказом Министерства здравоохранения РФ от 6 июня 2012 г. №4н «Об утверждении номенклатурной классификации медицинских изделий» и решением Коллегии ЕЭК №173 «Об утверждении Правил классификации медицинских изделий в зависимости от потенциального риска применения».
Цели и задачи клинического мониторинга направлены на:
- подтверждение качества клинической эффективности и безопасности медицинского изделия при применении в соответствии с назначением, определенным производителем;
- выявление ранее не учтенных рисков с учетом имеющихся клинических данных, специфических особенностей и факторов риска, связанных с медицинским изделием. Не допускается в целях и задачах указывать только отслеживание и анализ неблагоприятных событий.
Безопасность и эффективность изделия связаны соотношением "риск/польза".
Оценка рисков производится на основании информации о неблагоприятных событиях, а польза подтверждается достижением клинического эффекта от предназначенного применения изделия. При этом в плане клинического мониторинга должны быть определены критерии достижения приемлемых результатов применения изделия по назначению.
Клиническая эффективность подтверждается получением и статистическим анализом клинических данных по критериям, определенным в плане мониторинга в соответствии с целями и задачами и схемой мониторинга. Отсутствие неблагоприятных событий не может служить подтверждением эффективности изделия, которая доказывается только практикой клинического применения конкретного изделия:
- Для медицинских изделий, зарегистрированных по национальным правилам, план и схема пострегистрационного клинического мониторинга предоставляется с первоначальным отчетом.
- Для медицинских изделий, зарегистрированных по правилам ЕАЭС, проводится в соответствии с планом, представленным в комплекте регистрационных документов.
В случае выявления и подтверждения неблагоприятных событий или причинения вреда жизни и здоровью производитель программного медицинского изделия (ПМИ) обязан выполнить ряд превентивных мероприятий, предусмотренных приказом Минздрава №980н, включая информирование потребителей. Если производитель не будет принимать необходимых мер по предотвращению угрозы здоровью, Росздравнадзор имеет право изъять данное ПМИ из обращения.